
Spectrum and peptide sequence are sufficient for top-tier mass spectrometry proteomics identification
The Tesorai team has updated the bioRxiv preprint describing the implementation of Tesorai Search, including detailed benchmarking of Tesorai against other leading software across multiple datasets.

Tesorai
Author
January 9, 2026

The Tesorai team has updated the bioRxiv preprint describing the implementation of Tesorai Search, including detailed benchmarking of Tesorai against other leading software across multiple datasets.
Read the paper: https://doi.org/10.1101/2024.08.19.606805
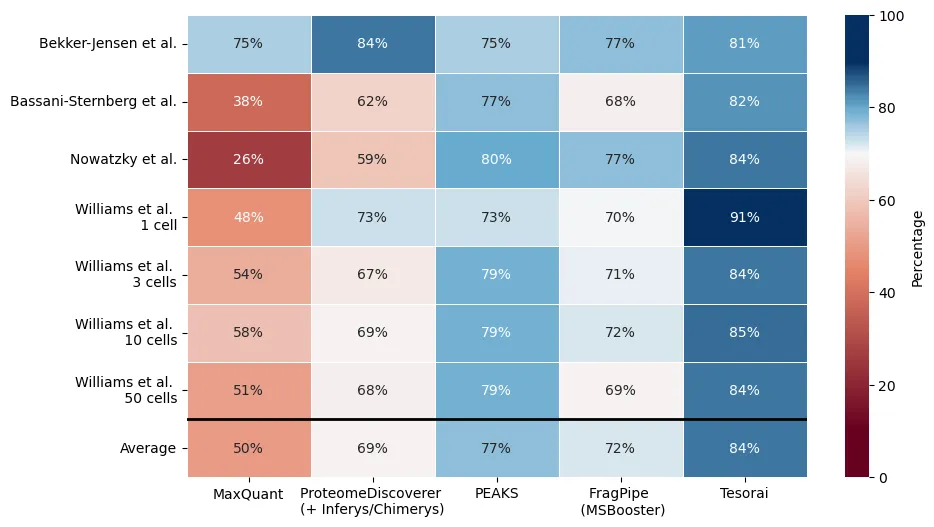
Abstract
The original mass spectrometry search engines used simple algorithms for peptide identification. Recent tools improved accuracy by adding several extra components such as fragment ion intensities or retention times prediction and training target-decoy classifiers on-the-fly, leading to sometimes inconsistent results.
Our study explores the impact of replacing those extra components with a deep-learning pretrained model that directly learns the complex relationship between the full spectra and associated peptide sequence, without using decoys. This simplified workflow has fewer parameters to tweak, making it easier to use and perform robustly on data from instruments and use-cases never seen during training.
Surprisingly, our approach consistently identifies more peptides than FragPipe, PEAKS, and Proteome Discoverer (12%, 9%, and 21% more, respectively, across a range of datasets). Tesorai Search is also fast – 250 immunopeptidomics searches in 45 minutes.
Resources
Explore publications, case studies, and guides to power your next experiment.

.svg)



We're here to help. Get in touch with our team or explore the platform yourself - and see how easy it is to get started
.svg)
Insights leveraging every byte of raw data